Neben der diagnostischen Tätigkeit in der täglichen Routine werden auch eine Vielfalt von Forschungsprojekten verfolgt und betreut.
Ein IZKF-gefördertes Projekt soll hier stellvertretend für die Forschung stehen.
Neben der diagnostischen Tätigkeit in der täglichen Routine werden auch eine Vielfalt von Forschungsprojekten verfolgt und betreut.
Ein IZKF-gefördertes Projekt soll hier stellvertretend für die Forschung stehen.
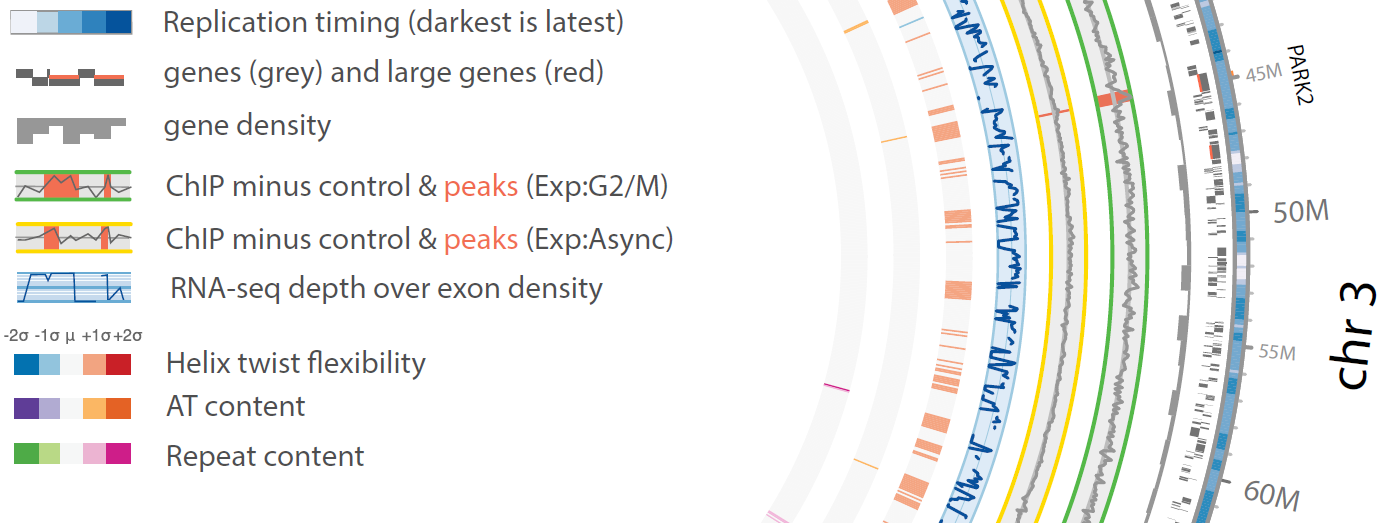
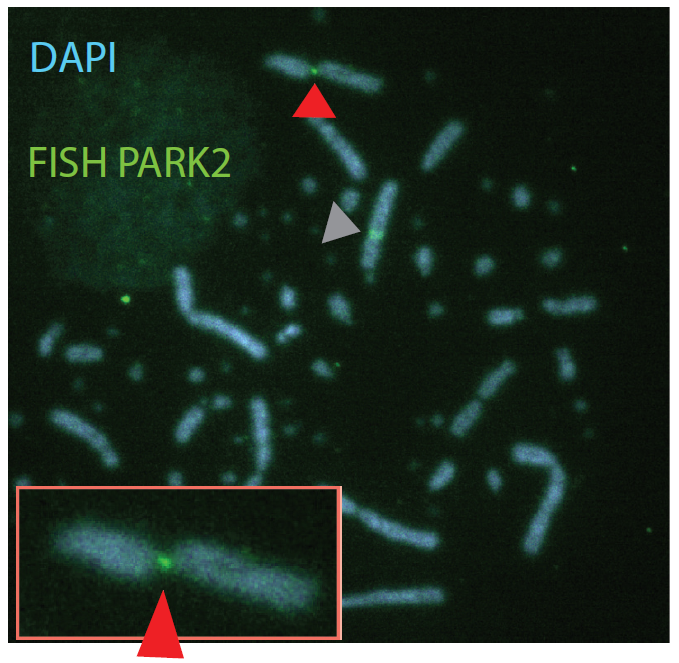
Chromosomal fragile sites (CFSs) are genomic regions prone to form gaps or breaks on metaphase chromosomes. As CFSs often appear as hot spots for gross chromosomal rearrangements in cancer cells, it is of high clinical relevance to map CFSs genome-wide for better understanding of complex aberrant karyotypes and to predict their clinical prognosis. For now, CFSs have mainly been analysed in human lymphocytes but the method is also established for other tissues and model systems. The mapping usually followed the traditional method by metaphase spreading and molecular cytogenetic definition. A new approach has become available recently to standardize this mapping to a high throughput and high resolution Next Generation Sequencing (NGS)-based genome-wide application. The aim of my junior research project is to establish this novel method in human cells, implement it for tumor material to overcome conventional metaphase spreading and morphological definition by a state-of-the-art NGS approach. To expand the view on CFSs by specific functional analyses in disease-derived cell lines, aspects of nuclear localization as well as molecular positioning of CFSs will be investigated. Overall, the project aims to identify the connection between the fragility of certain genomic regions and cancer predisposing and premature aging related disorders.