Hereditäre Sensomotorische Neuropathien (HMSN)
Hereditäre Sensomotorische Neuropathien (HMSN)
| Subtyp | Gensymbol | OMIM-Eintrag |
| CMT1A/HNPP | PMP22*,° | |
| CMT1B | MPZ° | OMIM 118200 |
| CMTX1 | GJB1° | OMIM 302800 |
| CMT2A2 | MFN2° | OMIM 609260 |
*Duplikations-/Deletionsanalyse mittels MLPA-Kit
°Direktsequenzierung
|
Methode: |
Next-Generation-Sequenzierung (NGS) Multiplex ligation-dependent probe amplification (MLPA) |
|
Material: |
2 - 5 ml EDTA-Blut oder 500 ng isolierte DNA |
|
Dauer: |
ca. 8 Wochen |
|
untersuchte Gene: |
siehe Anforderungsschein |
Hintergrund
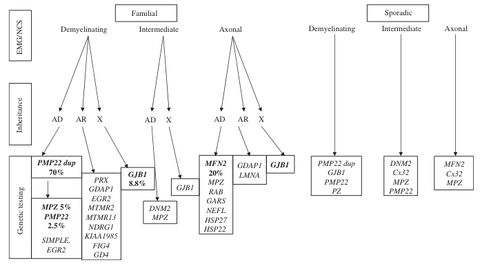
Es handelt sich bei der HMSN um eine genetisch sehr heterogene, erbliche Erkrankung des Nervensystems, die sowohl die motorischen Fähigkeiten (Bewegung) als auch die sensorischen Fähigkeiten (Empfindung) beeinflussen kann. Die Erkrankung wird nach Ihren Entdeckern auch Charcot-Marie-Tooth (CMT) genannt. Die HMSN ist eine Erkrankung der peripheren Nerven, bei der die isolierende Schicht um die Nerven (Myelinschicht) und/oder die Nervenfasern selbst zugrunde gehen. Charakteristisch ist eine symmetrische, distal betonte Muskelschwäche bzw. - atrophie. Meistens sind zunächst die Fuß- und Wadenmuskulatur betroffen, später auch die Handmuskeln. Fußdeformitäten wie Hohlfüße und Krallenzehen gehören zum klinischen Bild. Die sensiblen Ausfälle sind im Allgemeinen geringer ausgeprägt. In den meisten Fällen ist die Erkrankung langsam progredient, mit sehr variablem Verlauf. In den meisten Fällen folgt die HMSN einem autosomal dominanten Erbgang. Dies gilt sowohl für die HMSN I (demyelinisierender Typ) als auch für die HMSN II (axonaler Typ). Die X-chromosomale Vererbung ist bei der HMSN ebenfalls nicht selten. Insgesamt liegt molekulargenetisch am Häufigsten eine Duplikation des PMP22-Gens zugrunde. Die entsprechende Deletion des PMP22-Gens führt zu einer Neuropathie mit Neigung zu Druckläsionen.
Vorgeschlagenes diagnostisches Vorgehen aus:
Szigeti K, Lupski JR., Eur J Hum Genet. 2009 Jun;17(6):703-10
Ansprechpartner:

Arbeitsgruppenleiterin